

FDA Issues Guidance with Intent to Delay Enforcement of Law — A Complex Compliance Challenge: Modernization of Cosmetics Regulation Act of 2022 (MoCRA)
By Debra Rade |
One could almost hear a collective sigh of relief in the cosmetic industry on November 8, 2023. That day, the ...
Halloween Safety Tips 2021
By Debra Rade |
Halloween is approaching. The enthusiasm for it grows each year, at least in my neighborhood. Pretty scary out there! No ...
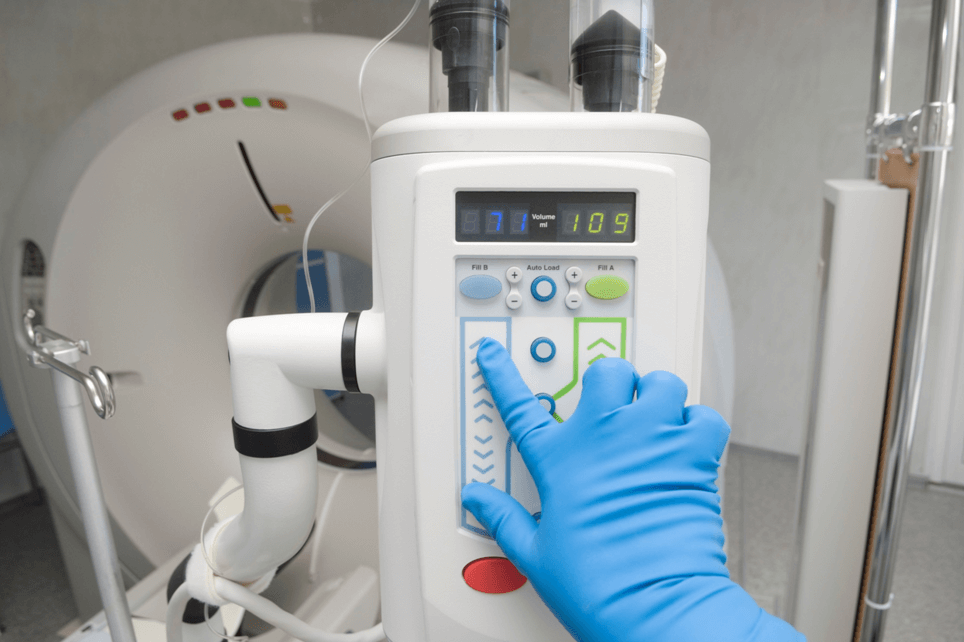
Access to Unique Device Identification
By Debra Rade |
There is a new website in town relevant to all medical devices called AccessGUDID. You decide if it is a catchy, ...

Baby Wipes, the FDA, and Your Baby’s Bottom
By Debra Rade |
With many friends who have delivered babies recently or who have been blessed with new grandchildren, it seems a good ...

The SEC Set Smith & Wesson in its Sight – FCPA Violations
By Debra Rade |
When the business market is saturated with your products, what’s next? Look to the global market. That’s just what Smith & ...

Hummus – Struggling for a New Standard of Identity?
By Debra Rade |
According to a HuffPost Taste post last year, there are at least 21 ways to make a better hummus, and a ...